Pathway analysis with PhosR
8 June 2026
Source:vignettes/web/pathway_analysis.Rmd
pathway_analysis.RmdIntroduction
Most phosphoproteomic studies have adopted a phosphosite-level
analysis of the data. To enable phosphoproteomic data analysis at the
gene level, PhosR implements both site- and gene-centric
analyses for detecting changes in kinase activities and signalling
pathways through traditional enrichment analyses (over-representation or
rank-based gene set test, together referred to as‘1-dimensional
enrichment analysis’) as well as 2- and 3-dimensional analyses.
This vignette will perform gene-centric pathway enrichment analyses
on the normalised myotube phosphoproteomic dataset using both
over-representation and rank–based gene set tests and also provide an
example of how directPA can be used to test which kinases
are activated upon different stimulations in myotubes using
2-dimensional analyses Yang
et al. 2014.
Loading packages and data
First, we will load the PhosR package with few other packages will use for the demonstration purpose.
We will use RUV normalised L6 phosphopreteome data for demonstration of gene-centric pathway analysis. It contains phosphoproteome from three different treatment conditions: (1) AMPK agonist AICAR, (2) insulin (Ins), and (3) in combination (AICAR+Ins).
suppressPackageStartupMessages({
library(calibrate)
library(limma)
library(directPA)
library(org.Rn.eg.db)
library(reactome.db)
library(annotate)
library(PhosR)
})
data("PhosphoSitePlus")We will use the ppe_RUV matrix from batch_correction.
1-dimensional enrichment analysis
To enable enrichment analyses on both gene and phosphosite levels,
PhosR implements a simple method called phosCollapse which
reduces phosphosite level of information to the proteins for performing
downstream gene-centric analyses. We will utilise two functions,
pathwayOverrepresent and
pathwayRankBasedEnrichment, to demonstrate 1-dimensional
(over-representation and rank-based gene set test) gene-centric pathway
enrichment analysis respectively.
First, extract phosphosite information from the ppe object.
sites = paste(sapply(ppe@GeneSymbol, function(x)x),";",
sapply(ppe@Residue, function(x)x),
sapply(ppe@Site, function(x)x),
";", sep = "")Then fit a linear model for each phosphosite.
f <- gsub("_exp\\d", "", colnames(ppe))
X <- model.matrix(~ f - 1)
fit <- lmFit(ppe@assays@data$normalised, X)Extract top-differentially regulated phosphosites for each condition compared to basal.
table.AICAR <- topTable(eBayes(fit), number=Inf, coef = 1)
table.Ins <- topTable(eBayes(fit), number=Inf, coef = 3)
table.AICARIns <- topTable(eBayes(fit), number=Inf, coef = 2)
DE1.RUV <- c(sum(table.AICAR[,"adj.P.Val"] < 0.05), sum(table.Ins[,"adj.P.Val"] < 0.05), sum(table.AICARIns[,"adj.P.Val"] < 0.05))
# extract top-ranked phosphosites for each group comparison
contrast.matrix1 <- makeContrasts(fAICARIns-fIns, levels=X) # defining group comparisons
contrast.matrix2 <- makeContrasts(fAICARIns-fAICAR, levels=X) # defining group comparisons
fit1 <- contrasts.fit(fit, contrast.matrix1)
fit2 <- contrasts.fit(fit, contrast.matrix2)
table.AICARInsVSIns <- topTable(eBayes(fit1), number=Inf)
table.AICARInsVSAICAR <- topTable(eBayes(fit2), number=Inf)
DE2.RUV <- c(sum(table.AICARInsVSIns[,"adj.P.Val"] < 0.05), sum(table.AICARInsVSAICAR[,"adj.P.Val"] < 0.05))
o <- rownames(table.AICARInsVSIns)
Tc <- cbind(table.Ins[o,"logFC"], table.AICAR[o,"logFC"], table.AICARIns[o,"logFC"])
rownames(Tc) <- sites[match(o, rownames(ppe))]
rownames(Tc) <- gsub("(.*)(;[A-Z])([0-9]+)(;)", "\\1;\\3;", rownames(Tc))
colnames(Tc) <- c("Ins", "AICAR", "AICAR+Ins")Summarize phosphosite-level information to proteins for the downstream gene-centric analysis.
Prepare the Reactome annotation
pathways = as.list(reactomePATHID2EXTID)
path_names = as.list(reactomePATHID2NAME)
name_id = match(names(pathways), names(path_names))
names(pathways) = unlist(path_names)[name_id]
pathways = pathways[which(grepl("Rattus norvegicus", names(pathways), ignore.case = TRUE))]
pathways = lapply(pathways, function(path) {
gene_name = unname(getSYMBOL(path, data = "org.Rn.eg"))
toupper(unique(gene_name))
})Perform 1D gene-centric pathway analysis
path1 <- pathwayOverrepresent(geneSet, annotation=pathways,
universe = rownames(Tc.gene), alter = "greater")
path2 <- pathwayRankBasedEnrichment(Tc.gene[,1],
annotation=pathways,
alter = "greater")Next, we will compare enrichment of pathways (in negative log10 p-values) between the two 1-dimensional pathway enrichment analysis. On the scatter plot, the x-axis and y-axis refer to the p-values derived from the rank-based gene set test and over-representation test, respectively. We find several expected pathways, while these highly enriched pathways are largely in agreement between the two types of enrichment analyses.
lp1 <- -log10(as.numeric(path2[names(pathways),1]))
lp2 <- -log10(as.numeric(path1[names(pathways),1]))
plot(lp1, lp2, ylab="Overrepresentation (-log10 pvalue)", xlab="Rank-based enrichment (-log10 pvalue)", main="Comparison of 1D pathway analyses", xlim = c(0, 10))
# select highly enriched pathways
sel <- which(lp1 > 1.5 & lp2 > 0.9)
textxy(lp1[sel], lp2[sel], gsub("_", " ", gsub("REACTOME_", "", names(pathways)))[sel])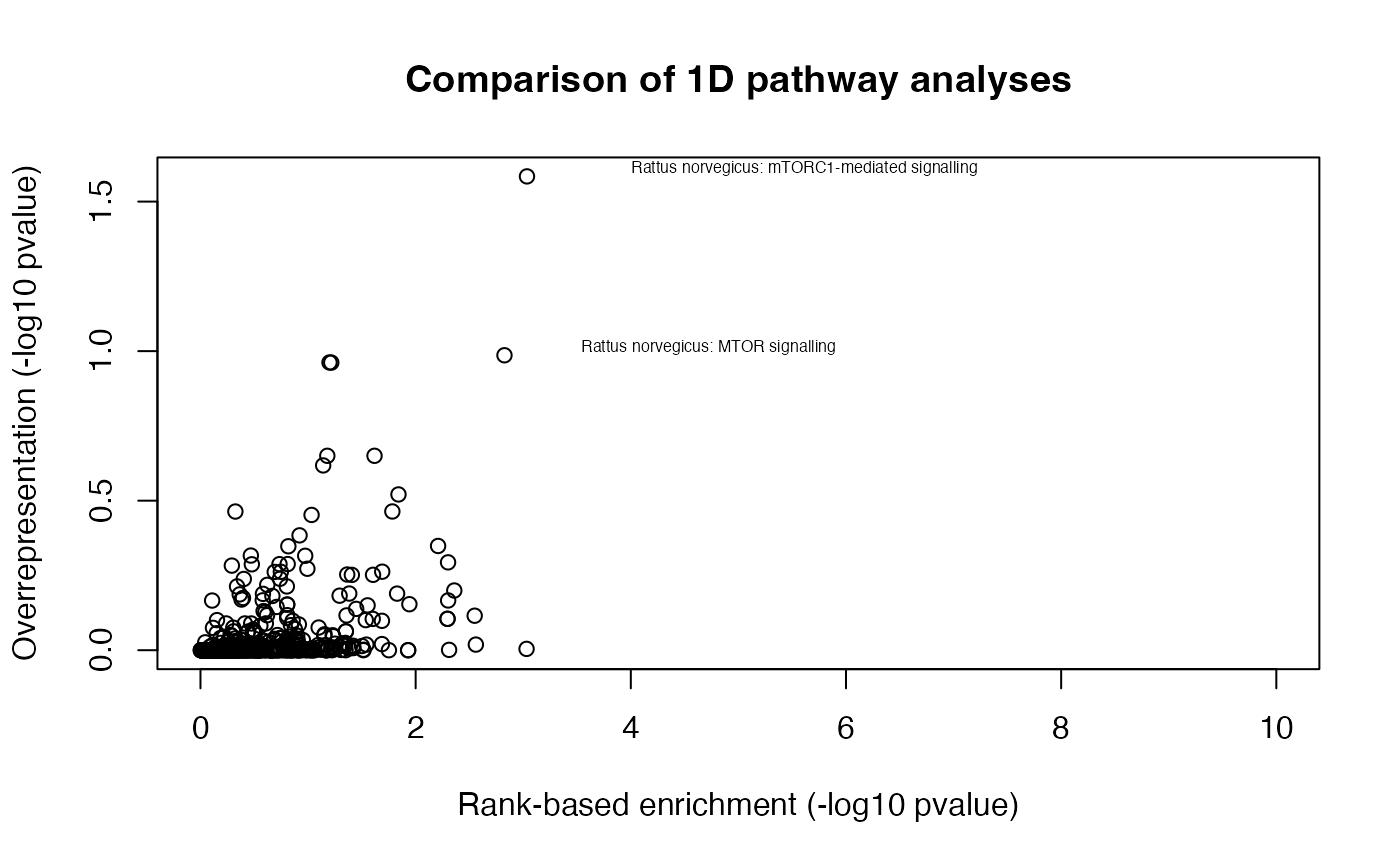
2- and 3-dimensional signalling pathway analysis
One key aspect in studying signalling pathways is to identify key
kinases that are involved in signalling cascades. To identify these
kinases, we make use of kinase-substrate annotation databases such as
PhosphoSitePlus and Phospho.ELM. These
databases are included in the PhosR and
directPA packages already. To access them, simply load the
package and access the data by data(“PhosphoSitePlus”) and
data(“PhosphoELM”).
The 2- and 3-dimensional analyses enable the investigation of kinases
regulated by different combinations of treatments. We will introduce
more advanced methods implemented in the R package directPA
for performing “2 and 3-dimentional” direction site-centric kinase
activity analyses.
# 2D direction site-centric kinase activity analyses
par(mfrow=c(1,2))
dpa1 <- directPA(Tc[,c(1,3)], direction=0,
annotation=lapply(PhosphoSite.rat, function(x){gsub(";[STY]", ";", x)}),
main="Direction pathway analysis")
dpa2 <- directPA(Tc[,c(1,3)], direction=pi*7/4,
annotation=lapply(PhosphoSite.rat, function(x){gsub(";[STY]", ";", x)}),
main="Direction pathway analysis")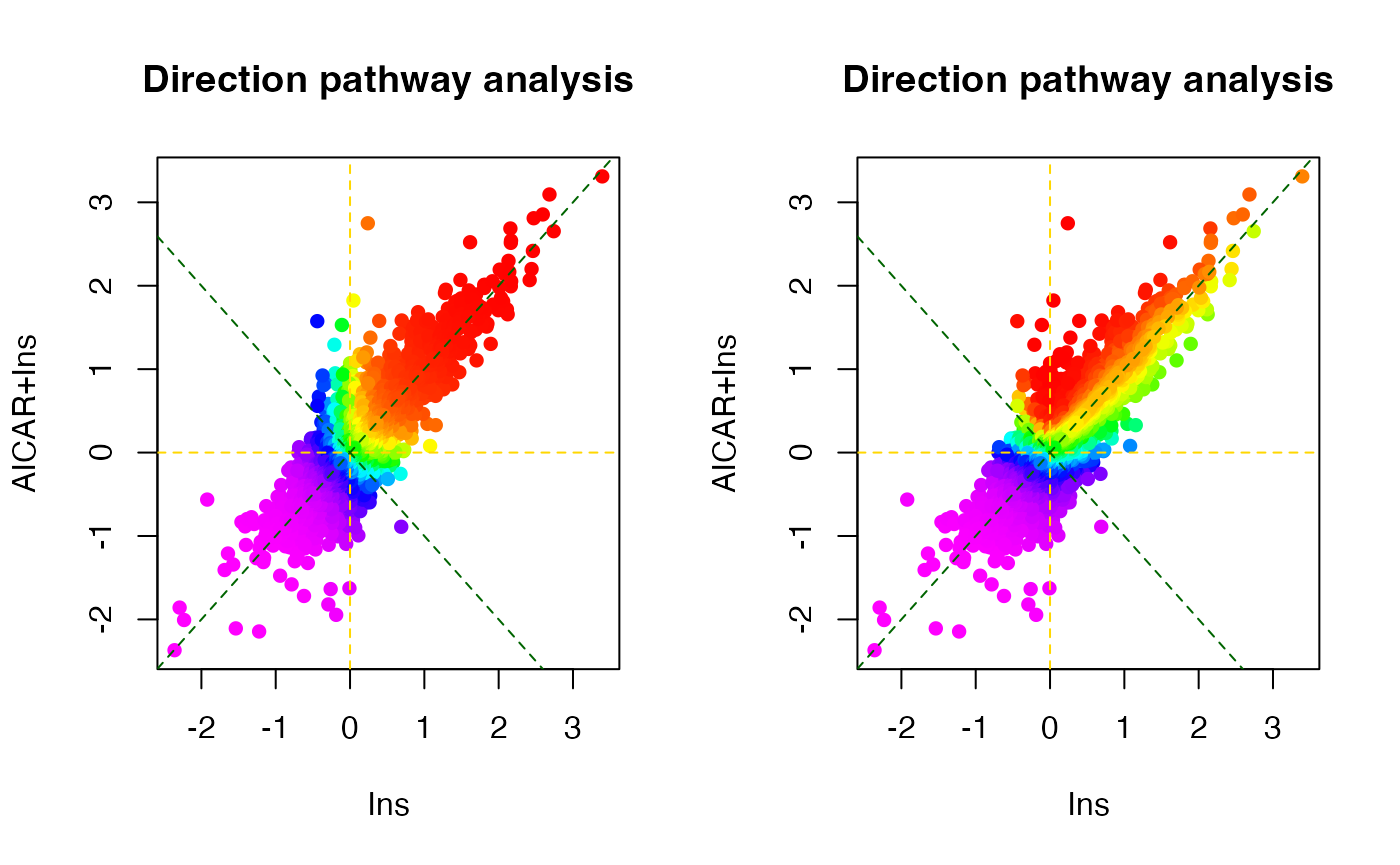
# top activated kinases
dpa1$pathways[1:5,]
#> pvalue size
#> AKT1 6.207001e-09 9
#> MAPK1 0.00057404 9
#> PRKACA 0.0006825021 25
#> PRKAA1 0.000965093 6
#> MAPK3 0.006670176 10
dpa2$pathways[1:5,]
#> pvalue size
#> PRKAA1 0.00463462 6
#> AKT1 0.02942273 9
#> CSNK2A1 0.2193148 12
#> CDK5 0.2607434 5
#> MAPK1 0.2767886 9There is also a function called perturbPlot2d
implemented in kinasePA for testing and visualising
activity of all kinases on all possible directions. Below are the
demonstration from using this function.
z1 <- perturbPlot2d(Tc=Tc[,c(1,2)],
annotation=lapply(PhosphoSite.rat, function(x){gsub(";[STY]", ";", x)}),
cex=1, xlim=c(-2, 4), ylim=c(-2, 4),
main="Kinase perturbation analysis")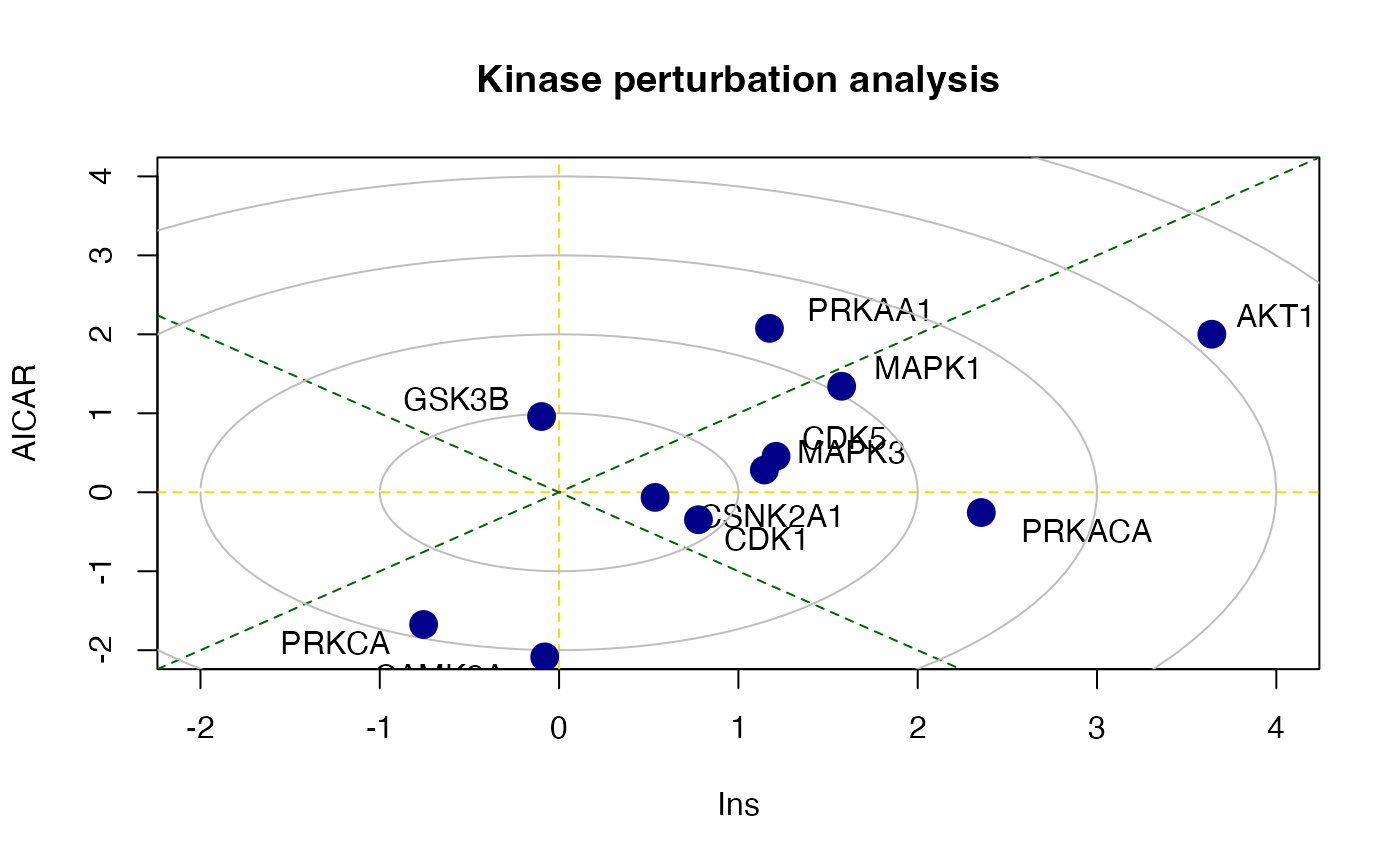
z2 <- perturbPlot2d(Tc=Tc[,c(1,3)], annotation=lapply(PhosphoSite.rat, function(x){gsub(";[STY]", ";", x)}),
cex=1, xlim=c(-2, 4), ylim=c(-2, 4),
main="Kinase perturbation analysis")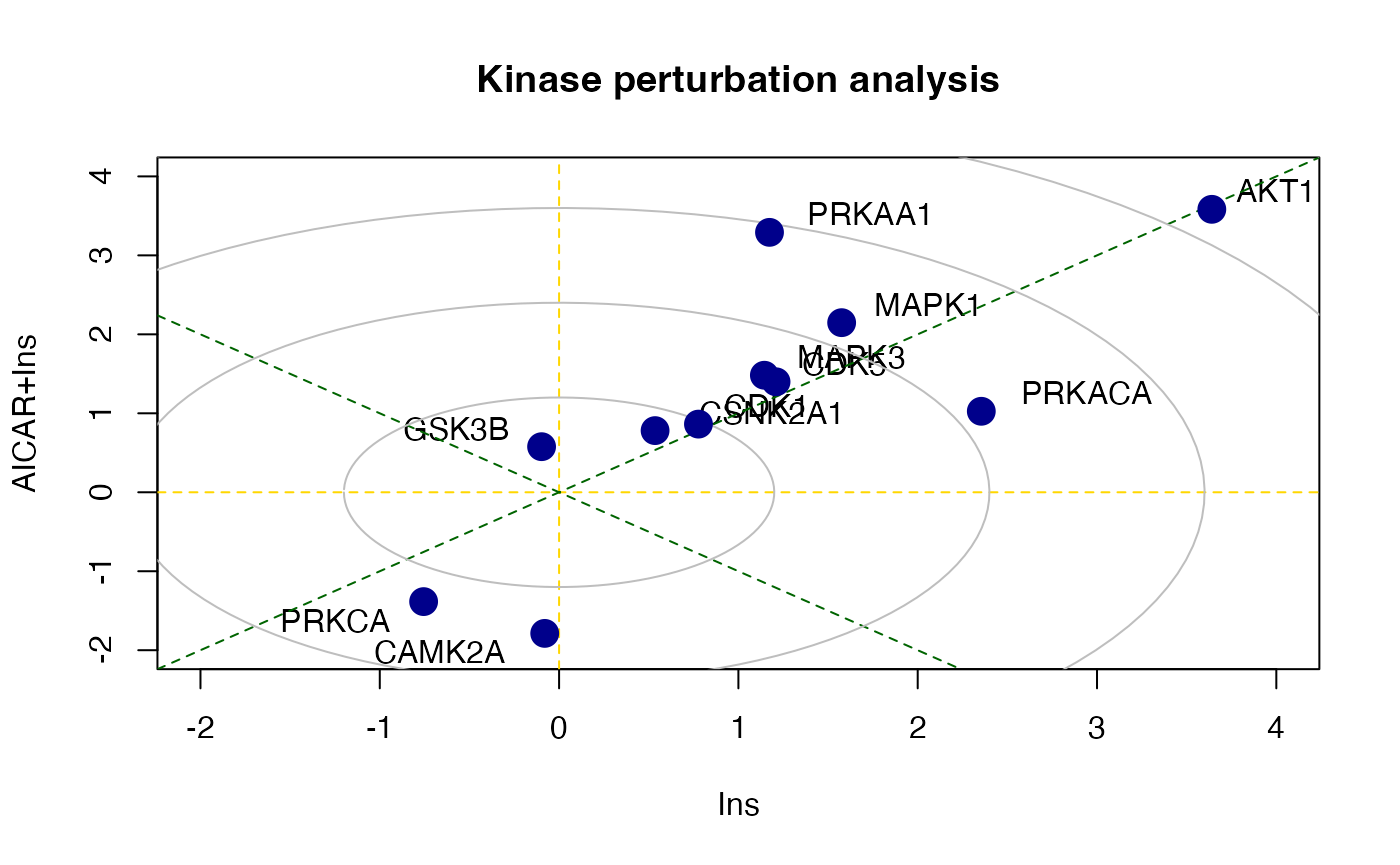
z3 <- perturbPlot2d(Tc=Tc[,c(2,3)], annotation=lapply(PhosphoSite.rat, function(x){gsub(";[STY]", ";", x)}),
cex=1, xlim=c(-2, 4), ylim=c(-2, 4),
main="Kinase perturbation analysis")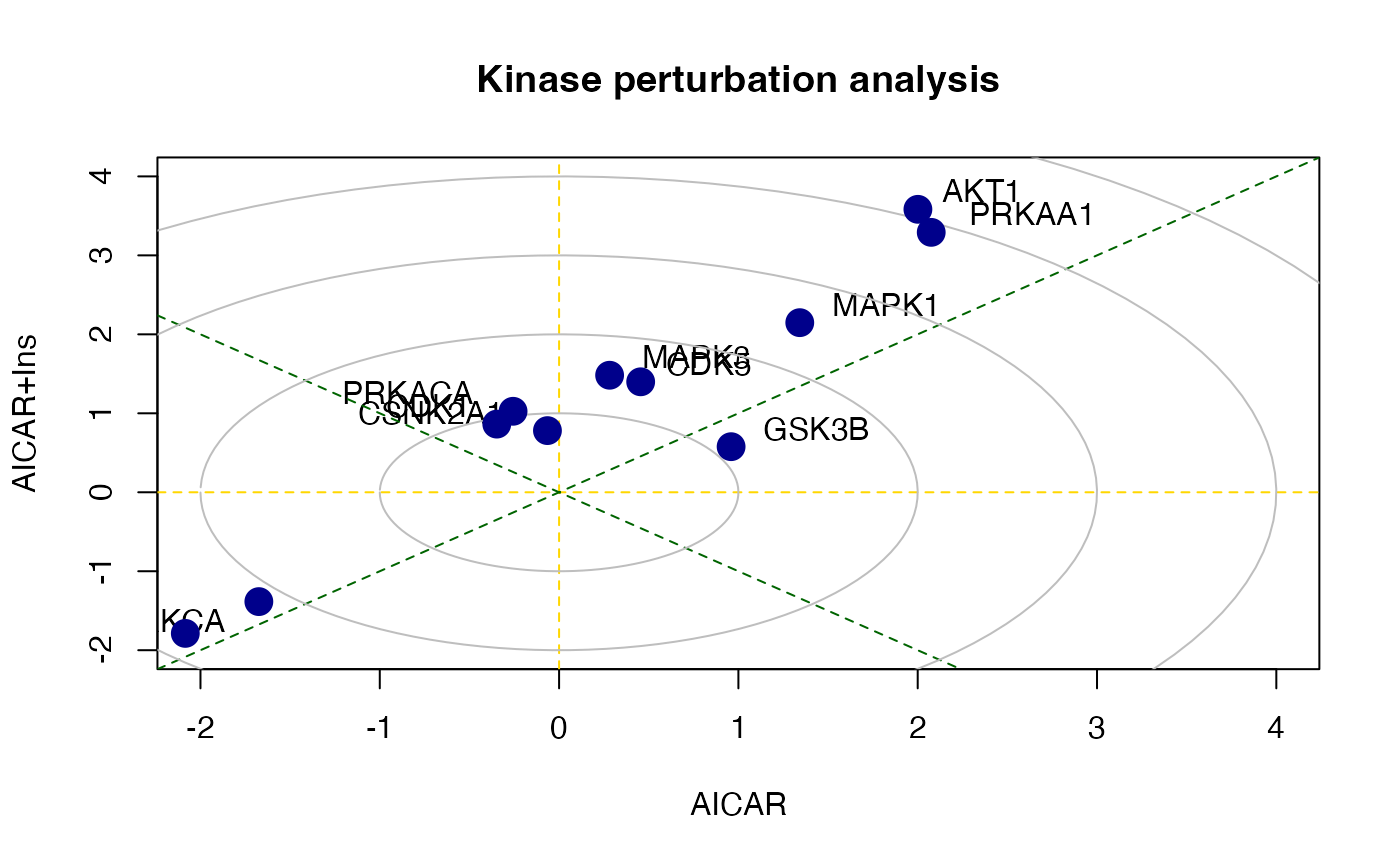
SessionInfo
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] PhosR_1.20.0 annotate_1.90.0 XML_3.99-0.23
#> [4] reactome.db_1.96.0 org.Rn.eg.db_3.23.0 AnnotationDbi_1.74.0
#> [7] IRanges_2.46.0 S4Vectors_0.50.1 Biobase_2.72.0
#> [10] BiocGenerics_0.58.1 generics_0.1.4 directPA_1.5.1
#> [13] limma_3.68.4 calibrate_1.7.7 MASS_7.3-65
#>
#> loaded via a namespace (and not attached):
#> [1] DBI_1.3.0 gridExtra_2.3
#> [3] rlang_1.2.0 magrittr_2.0.5
#> [5] otel_0.2.0 matrixStats_1.5.0
#> [7] e1071_1.7-17 compiler_4.6.0
#> [9] RSQLite_3.53.1 reshape2_1.4.5
#> [11] png_0.1-9 systemfonts_1.3.2
#> [13] vctrs_0.7.3 stringr_1.6.0
#> [15] pkgconfig_2.0.3 shape_1.4.6.1
#> [17] crayon_1.5.3 fastmap_1.2.0
#> [19] backports_1.5.1 XVector_0.52.0
#> [21] rmarkdown_2.31 preprocessCore_1.74.0
#> [23] ragg_1.5.2 network_1.20.0
#> [25] purrr_1.2.2 bit_4.6.0
#> [27] xfun_0.58 cachem_1.1.0
#> [29] jsonlite_2.0.0 blob_1.3.0
#> [31] DelayedArray_0.38.2 broom_1.0.13
#> [33] R6_2.6.1 stringi_1.8.7
#> [35] bslib_0.11.0 RColorBrewer_1.1-3
#> [37] GGally_2.4.0 car_3.1-5
#> [39] GenomicRanges_1.64.0 jquerylib_0.1.4
#> [41] Rcpp_1.1.1-1.1 Seqinfo_1.2.0
#> [43] SummarizedExperiment_1.42.0 knitr_1.51
#> [45] igraph_2.3.2 Matrix_1.7-5
#> [47] tidyselect_1.2.1 abind_1.4-8
#> [49] yaml_2.3.12 viridis_0.6.5
#> [51] plyr_1.8.9 lattice_0.22-9
#> [53] tibble_3.3.1 KEGGREST_1.52.0
#> [55] S7_0.2.2 coda_0.19-4.1
#> [57] evaluate_1.0.5 desc_1.4.3
#> [59] ggstats_0.13.0 proxy_0.4-29
#> [61] circlize_0.4.18 Biostrings_2.80.1
#> [63] pillar_1.11.1 BiocManager_1.30.27
#> [65] ggpubr_0.6.3 carData_3.0-6
#> [67] MatrixGenerics_1.24.0 ggplot2_4.0.3
#> [69] scales_1.4.0 BiocStyle_2.40.0
#> [71] xtable_1.8-8 class_7.3-23
#> [73] glue_1.8.1 pheatmap_1.0.13
#> [75] tools_4.6.0 dendextend_1.19.1
#> [77] ggsignif_0.6.4 fs_2.1.0
#> [79] grid_4.6.0 tidyr_1.3.2
#> [81] colorspace_2.1-2 Formula_1.2-5
#> [83] cli_3.6.6 ruv_0.9.7.1
#> [85] textshaping_1.0.5 S4Arrays_1.12.0
#> [87] viridisLite_0.4.3 ggdendro_0.2.0
#> [89] dplyr_1.2.1 pcaMethods_2.4.0
#> [91] gtable_0.3.6 rstatix_0.7.3
#> [93] sass_0.4.10 digest_0.6.39
#> [95] SparseArray_1.12.2 htmlwidgets_1.6.4
#> [97] farver_2.1.2 memoise_2.0.1
#> [99] htmltools_0.5.9 pkgdown_2.2.0
#> [101] lifecycle_1.0.5 httr_1.4.8
#> [103] statnet.common_4.13.0 GlobalOptions_0.1.4
#> [105] statmod_1.5.2 bit64_4.8.2