Quickstart¶
One model, four tasks
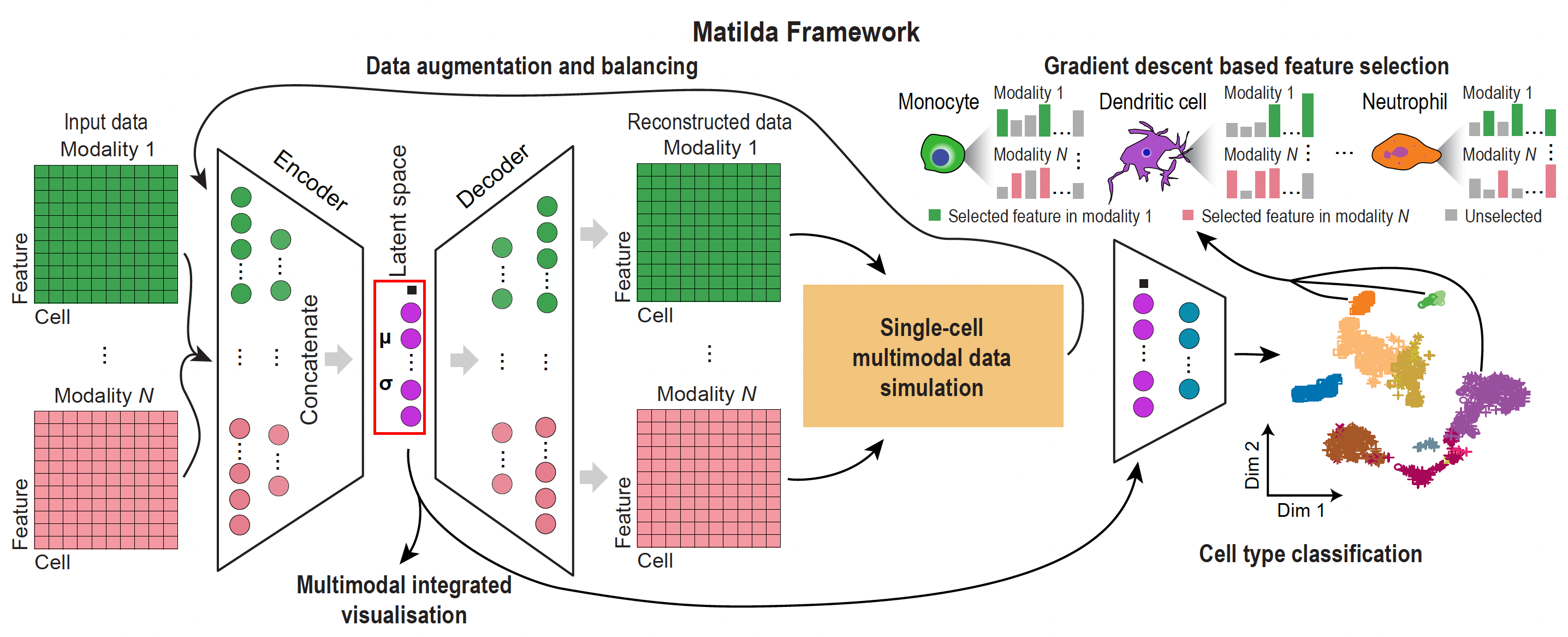
Matilda trains one multimodal VAE + classifier and reuses it for classification,
dimension reduction, feature selection, and data simulation. The two interfaces below (the
Python matilda-sc object API and the R object API) drive the same model and give the
same results. See Installation to set up either one.
End to end in five lines¶
The example below is self-contained: it downloads the TEA-seq (RNA + ADT + ATAC) demo
(~75 MB, cached after the first run), then runs the full workflow — train → reduce → classify →
markers → simulate. Copy-paste and run. In Python you pass in-memory AnnData (one per
modality) to matilda.train() then the task verbs; in R you pass a SingleCellExperiment (SCE)
and let the model ride along inside the object.
import matilda
from matilda import io
import os, urllib.request, tarfile, pandas as pd
# demo data: download + cache the TEA-seq demo (~75 MB, first run only)
DATA = os.path.expanduser("~/.cache/matilda/matilda_teaseq_demo")
if not os.path.isdir(DATA):
url = "https://github.com/PYangLab/Matilda/releases/download/demo-data/matilda_teaseq_demo.tar.gz"
os.makedirs(os.path.dirname(DATA), exist_ok=True)
tgz, _ = urllib.request.urlretrieve(url)
tarfile.open(tgz).extractall(os.path.dirname(DATA))
rna, adt, atac = (io.read_matilda_h5(f"{DATA}/train_{m}.h5") for m in ("rna", "adt", "atac")) # reference
q_rna, q_adt, q_atac = (io.read_matilda_h5(f"{DATA}/test_{m}.h5") for m in ("rna", "adt", "atac")) # held-out query
labels = pd.read_csv(f"{DATA}/train_cty.csv", index_col=0).iloc[:, 0].values
fit = matilda.train(rna, adt, atac, labels=labels) # 1. train (one shared model)
red = matilda.reduce({"rna": rna, "adt": adt, "atac": atac}, model=fit) # 2. latent space
res = matilda.classify({"rna": q_rna, "adt": q_adt, "atac": q_atac}, model=fit) # 3. cell types
mk = matilda.markers({"rna": rna, "adt": adt, "atac": atac}, model=fit, labels=labels) # 4. markers
sim = matilda.simulate({"rna": rna, "adt": adt, "atac": atac}, model=fit,
celltype="B.Naive", n=200, labels=labels) # 5. synthetic cells
library(matilda)
library(SingleCellExperiment)
# demo data: download + cache the TEA-seq demo (~75 MB, first run only)
dir <- matilda_download_example()
read_h5 <- function(path) { # native Matilda .h5 -> features x cells
m <- rhdf5::h5read(path, "matrix/data")
feats <- as.character(rhdf5::h5read(path, "matrix/features"))
cells <- as.character(rhdf5::h5read(path, "matrix/barcodes"))
if (nrow(m) == length(cells) && ncol(m) == length(feats)) m <- t(m)
dimnames(m) <- list(feats, cells); m
}
make_sce <- function(split) { # build an SCE for "train" or "test"
h5 <- function(mod) file.path(dir, sprintf("%s_%s.h5", split, mod))
sce <- SingleCellExperiment(assays = list(counts = read_h5(h5("rna"))))
altExp(sce, "ADT") <- SummarizedExperiment(list(counts = read_h5(h5("adt"))))
altExp(sce, "ATAC") <- SummarizedExperiment(list(counts = read_h5(h5("atac"))))
sce$cell_type <- as.character(read.csv(file.path(dir, sprintf("%s_cty.csv", split)), header = FALSE)[[2]])[-1]
sce
}
sce <- make_sce("train") # reference
query <- make_sce("test") # held-out query
sce <- matilda_train(sce, label = "cell_type") # 1. train (model stored in the object)
sce <- matilda_reduce(sce) # 2. reducedDim "MATILDA"
query <- matilda_classify(query, reference = sce) # 3. colData$matilda_pred / $matilda_prob
mk <- matilda_markers(sce) # 4. per-cell-type feature importance
sim <- matilda_simulate(sce, celltype = "B.Naive", n = 200) # 5. synthetic cells
What each step does¶
| Step | Python | R | Result |
|---|---|---|---|
| Train | matilda.train(rna, adt, atac, labels=) |
matilda_train(sce, label=) |
one shared model |
| Reduce | matilda.reduce(data, model=) |
matilda_reduce(sce) |
integrated latent space (reducedDim "MATILDA") |
| Classify | matilda.classify(query, model=) |
matilda_classify(query, reference=) |
predicted cell types (colData$matilda_pred) |
| Markers | matilda.markers(data, model=) |
matilda_markers(sce) |
per-cell-type feature importance |
| Simulate | matilda.simulate(data, model=, celltype=, n=) |
matilda_simulate(sce, celltype=, n=) |
synthetic cells |
The trained model is carried as model= in Python and reference= in R. classify reconciles
features automatically: it reuses the model when the query shares the reference panel, and
retrains on the reference ∩ query intersection when it doesn't (real values, no zero-padding).
Hyperparameters (batch_size, epochs, lr, z_dim, hidden_rna, hidden_adt, hidden_atac,
seed, augmentation) default to the published settings and are shared verbatim across both APIs.
Next steps¶
- Full Python walkthrough on TEA-seq: Python tutorial
- Full R walkthrough on TEA-seq: R tutorial
- Setting up either interface: Installation